This tutorial shows how to use the Infernal software to annotate the reference SARS-CoV-2 genome with RNA families from Rfam.
The same approach can be used to find RNA families in any RNA or DNA sequence.
- Docker Desktop on your computer in order to access a complete working environment pre-configured using Docker.
💡 Alternatively, try Play with Docker (PWD) in your browser (requires a free Docker account and depends on the resource availability).


Download a pre-built Docker image containing all data and software:
docker pull rfam/tutorials
Start an interactive session:
docker run -it rfam/tutorials
You should see a screen similar to the following:
$ docker run -it rfam/tutorials
rfam-user@48a963da2278:~$
You can now type any bash commands and follow the instructions below.
-
Type
lsto list files in your folder. You should see:Rfam.cm- Rfam covariance models from release 14.5Rfam.clanin- A list of Rfam clansvirus.fasta- SARS-CoV-2 sequence NC_045512.2
-
Run
cmpress Rfam.cmto prepare the Rfam covariance models to be used by Infernal (takes ~15 s, you only need to do this once). -
Run Infernal
cmscanto find Rfam families invirus.fasta(the command should take 30-60 seconds):cmscan --cut_ga --rfam --nohmmonly --clanin Rfam.clanin --oskip --fmt 2 -o output.txt --tblout table.txt Rfam.cm virus.fastaHere is a quick explanation of the command line options:
--cut_ga- use the thresholds selected by Rfam curators--rfam- run in “fast” mode, the same mode used for Rfam annotation--nohmmonly- run all models in CM mode (not HMM mode). This ensures all GA cutoffs, which were determined in CM mode for each model, are valid--clanin Rfam.clanin --fmt 2 --oskip- remove redundant hits from the same Rfam clan-o output.txt- cmscan output including alignments--tblout table.txt- cmscan output table
⚠️ It is recommended to always use the--cut_ga --rfam --nohmmonlyoptions when annotating genomes with Rfam families. -
Inspect the output files output.txt and table.txt:
less -S output.txt less -S table.txt -
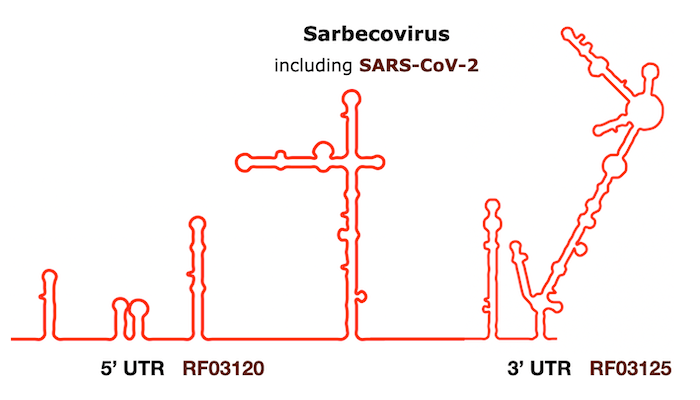
Find the Rfam families from the Infernal output on the figure from Huffsky et al., 2020:
-
Bonus points: repeat step 3 without the
--oskipoption. Notice the additional hits from the bCoV-5UTR and bCoV-3UTR families (see table-no-oskip.txt).

-
Download or clone this repository and move to the directory:
git clone https://github.com/Rfam/rfam-tutorials.git cd rfam-tutorials -
Build a docker image:
docker build -t rfam/tutorials . -
Start a docker container and mount the data folder:
docker run -v `pwd`/data:/home/rfam-user/data -it rfam/tutorials
- See Alternate Protocol 1 in Kalvari et al., 2018 for more details about annotating a genome with Infernal and Rfam
- Rfam SARS-CoV-2 annotations are described in Huffsky et al., 2020
- Find out about other Infernal commands in the Infernal User Guide
If you have any feedback, feel free to create an issue or submit a pull request.